Welcome to the COPD subtypes blog! This is the place to come for our most up-to-date thoughts about our approaches/findings regarding COPD subtypes. As of January 2020 we have five posts, which are summarized below. For those who want to start with a bigger picture summary, post #5 is the best place to start. Otherwise, if you go through them sequentially that more closely follows our published papers and the natural evolution of our groups’ thinking on subtypes.
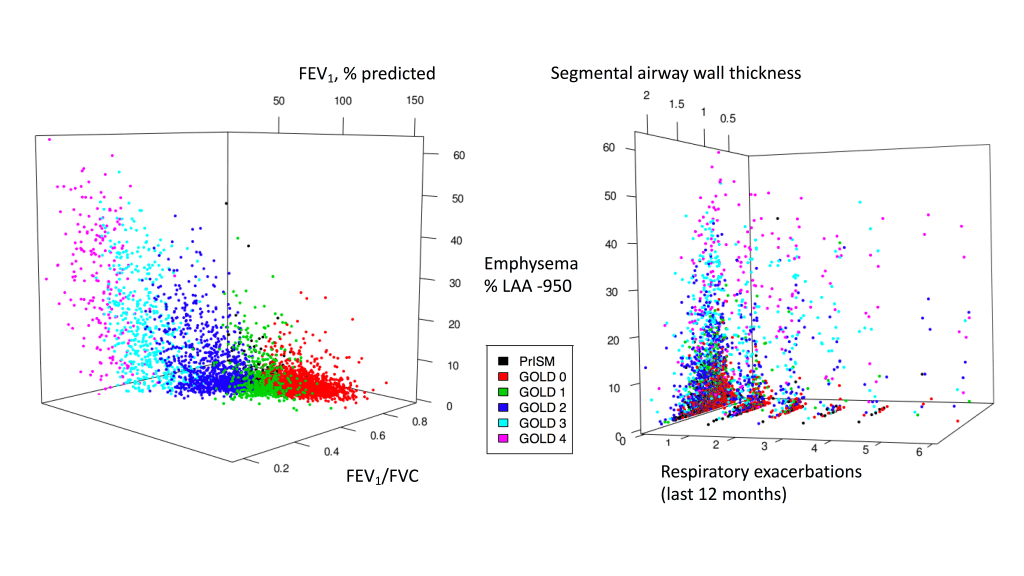
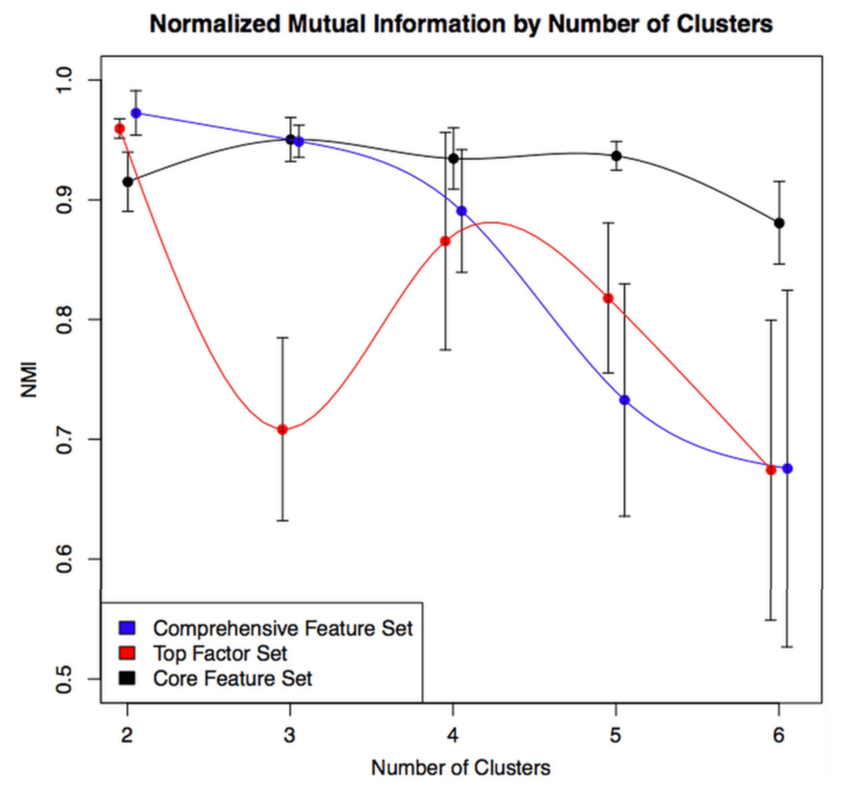
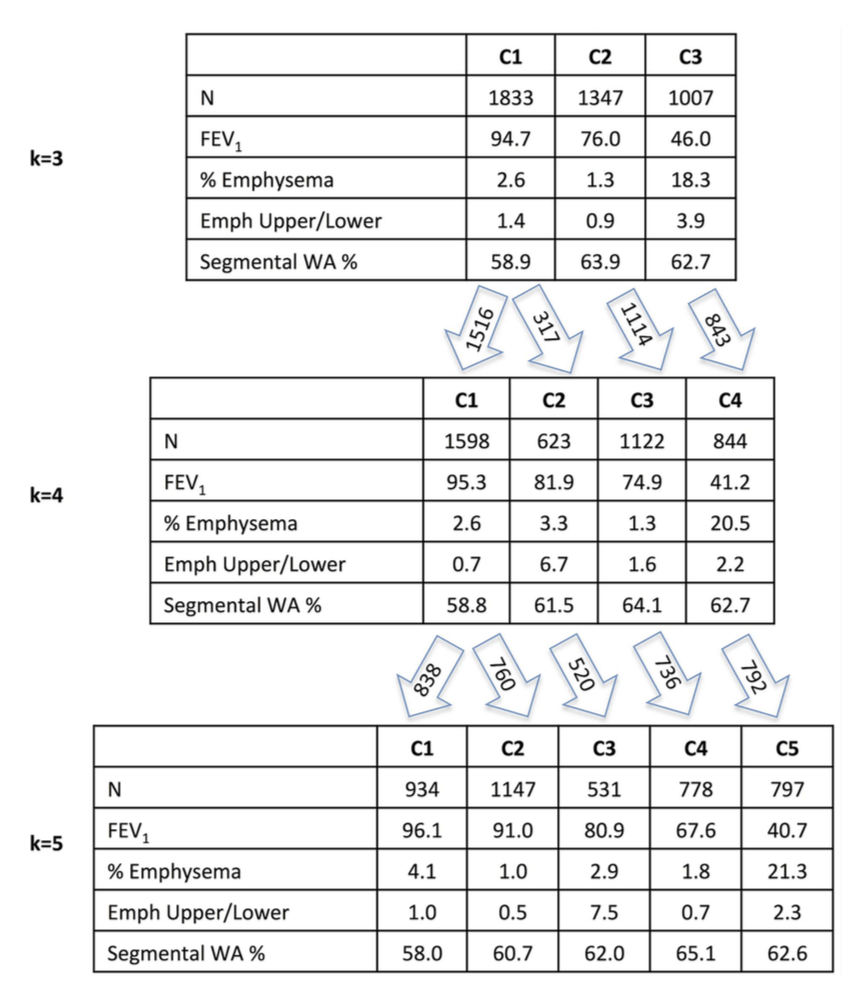
1: The COPD continuum and the downsides of clustering. Reviews one of our first subtyping papers and discusses the fact that COPD data usually do not have clear clustering structure.
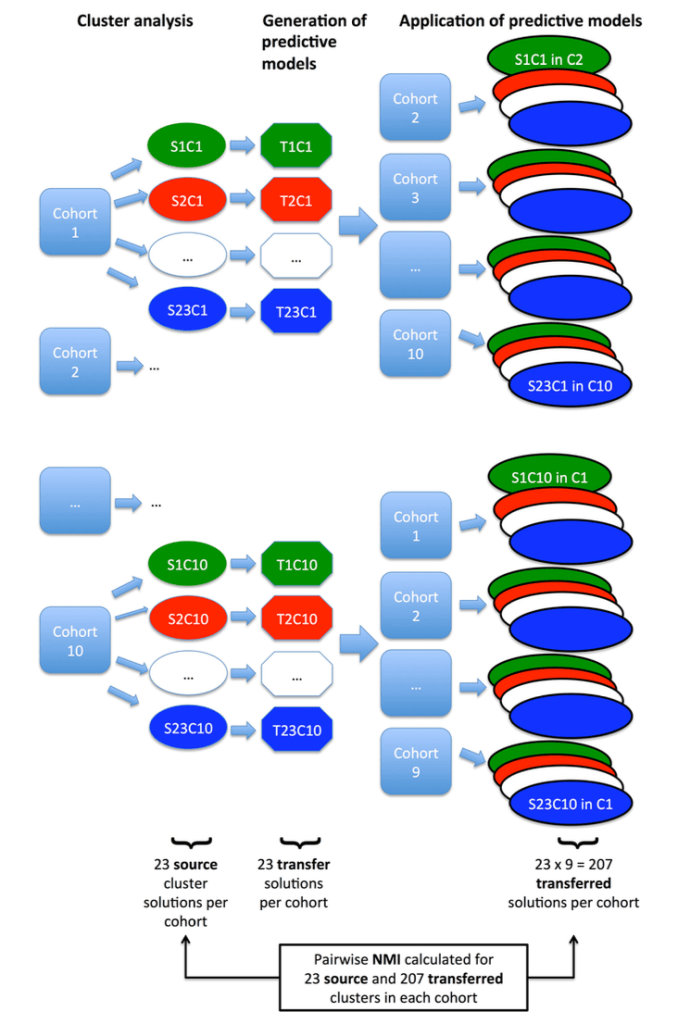
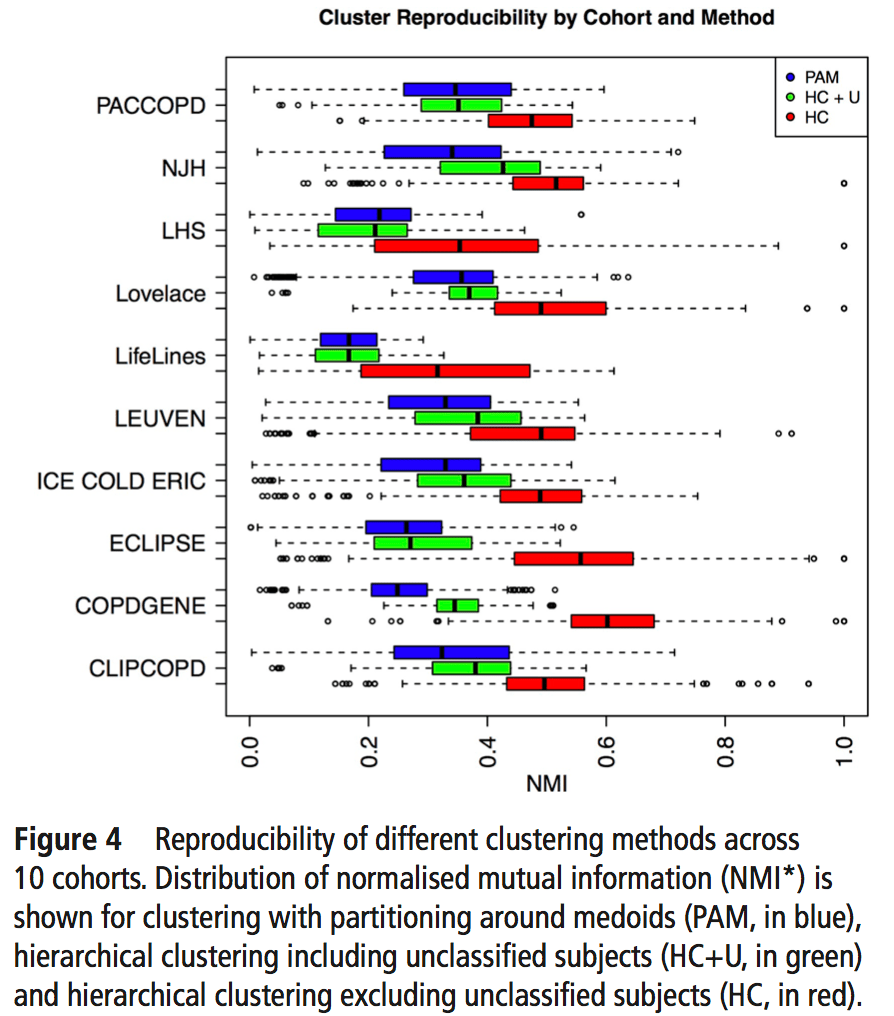
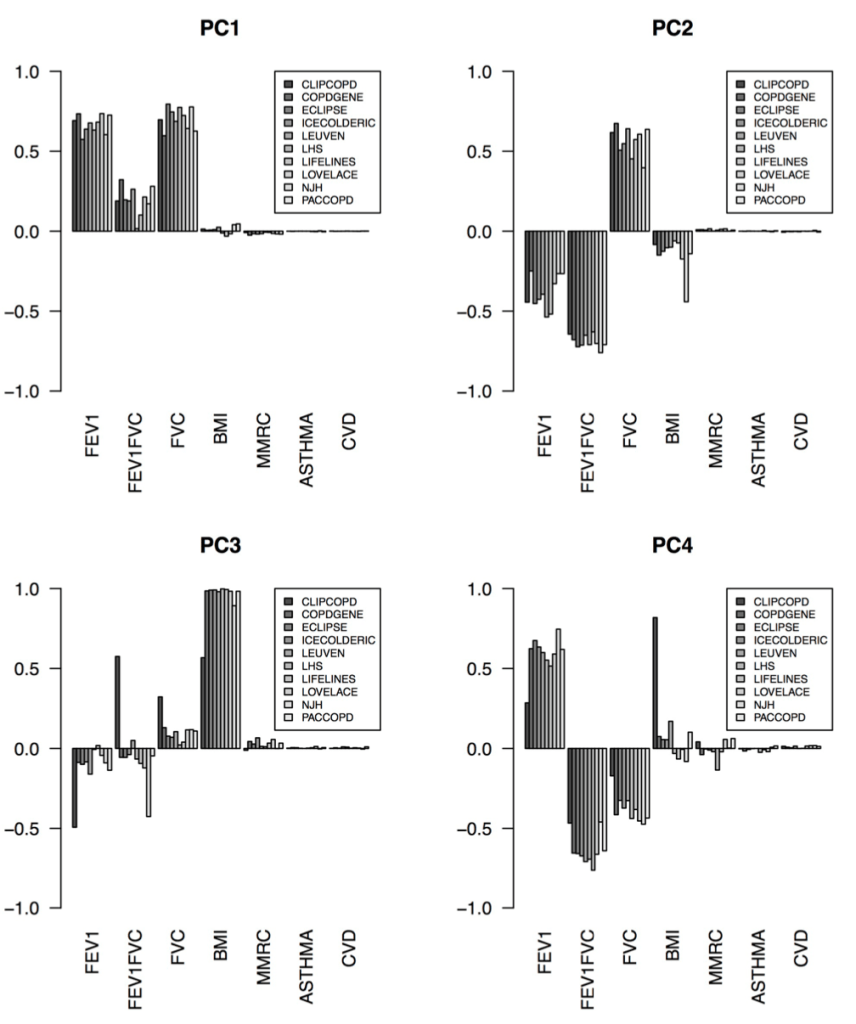
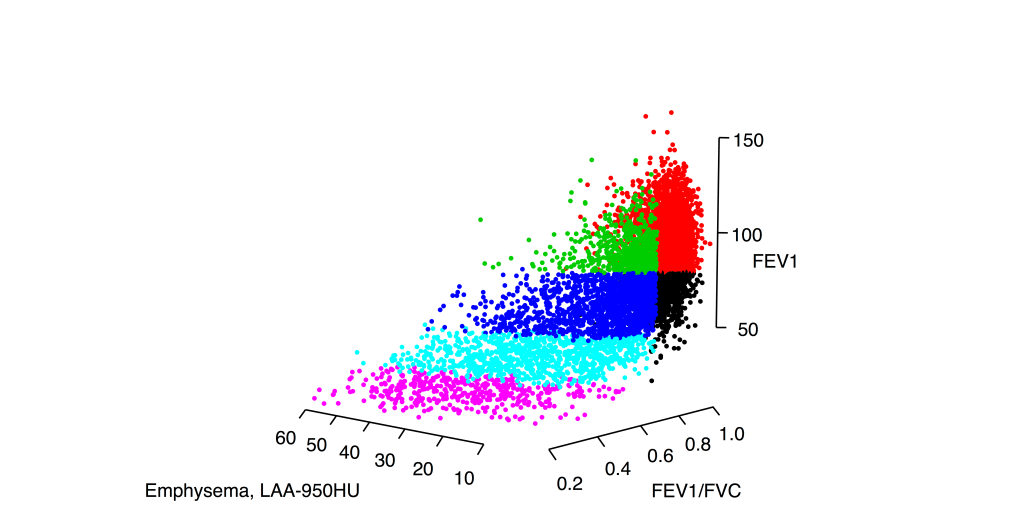
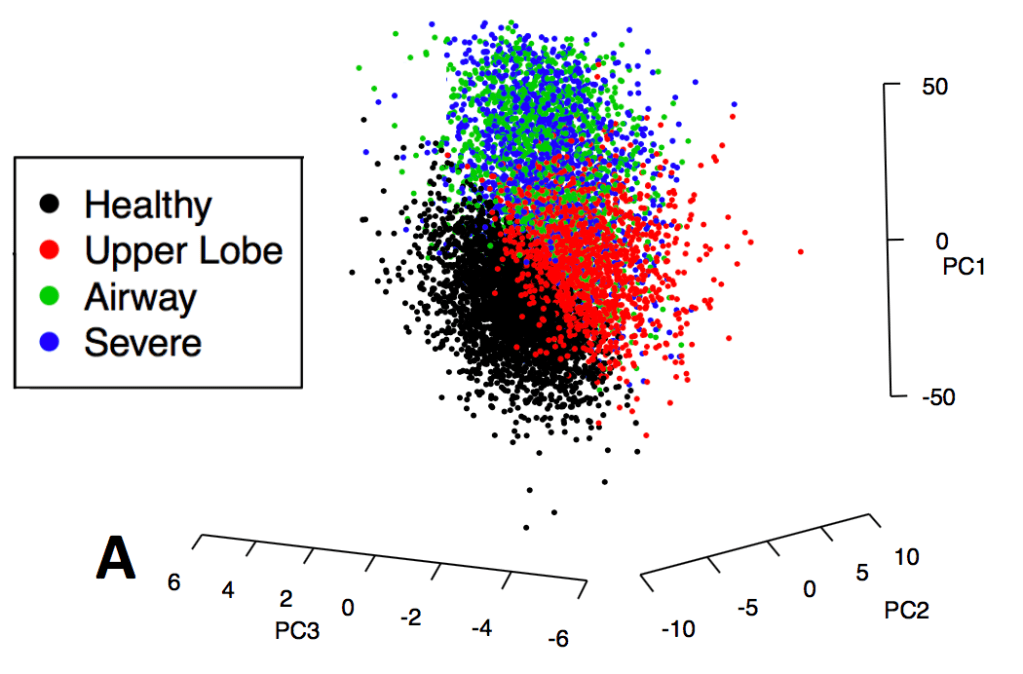
2: Reproducibility: Disease axes > clusters, a 10 cohort validation study. Reviews our collaborative paper that demonstrates 1) problems with clustering reproducibility in independent data and 2) high similarity of PCA axes across those same cohorts.
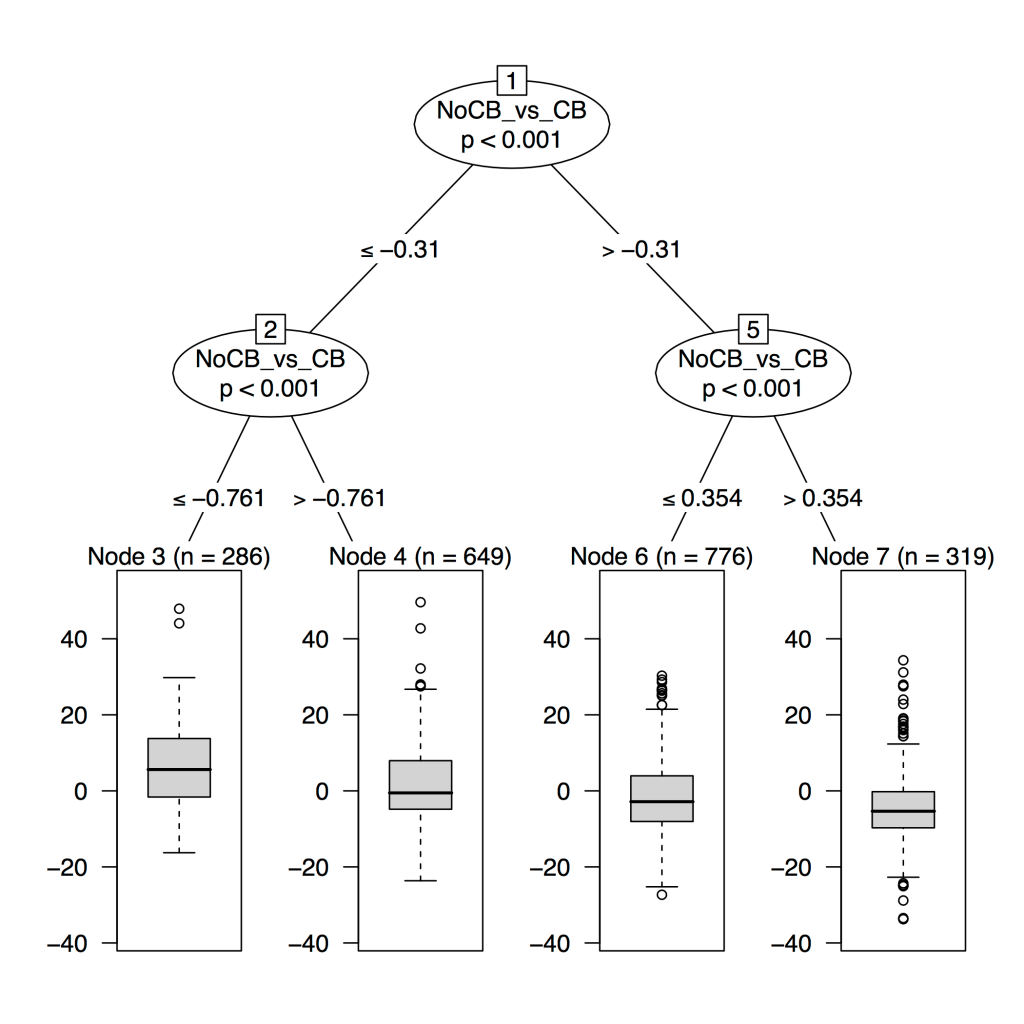
3: For COPD, think disease axes before clusters. A brief post that illustrates the importance of defining feature spaces (i.e. the variables that are most important for cluster identification) and introduces supervised learning and disease axes as a promising way to do this.
4: A clustering alternative: supervised prediction for disease axis discovery. Makes the case for an alternative path to clusters that goes first through supervised prediction and disease axes, then to outcome-driven subtypes.
5: What are COPD subtypes for? A higher level summary of how disease axes and supervised learning can be a useful approach.